Intestinal Organoid
This tutorial uses the intestine organoid data from Battich, et al (2020). Based on the original paper, the differentiation process happens from stem cells to two distinct branches, secretory and enterocyte.
In this tutorial, we will walk you through the workflow of UniTVelo analysis and utilizing regression \(R^2\) to identify genes contributes to branching.
Intestine organoid dataset has been used in other velocity calculation methods with the help of metabolic labelling data. Here with UniTVelo, only transcriptomics data are used as input.
[1]:
import scvelo as scv
scv.settings.verbosity = 0
import unitvelo as utv
(Running UniTVelo 0.1.0)
2022-03-31 14:22:17
[7]:
dataset = '../data/Organoids/organoids.h5ad'
label = 'cell_type'
exp_metrics = {}
[8]:
adata = scv.read(dataset)
adata
[8]:
AnnData object with n_obs × n_vars = 3831 × 9157
obs: 'well_id', 'batch_id', 'treatment_id', 'log10_gfp', 'som_cluster_id', 'monocle_branch_id', 'monocle_pseudotime', 'rotated_umap1', 'rotated_umap2', 'cell_type'
var: 'gene'
obsm: 'X_umap'
layers: 'spliced', 'unspliced'
[9]:
cluster_edges = [
("Stem cells", "TA cells"),
("Stem cells", "Goblet cells")]
[10]:
adata = scv.read(dataset)
cell_mapper = {
'1': 'Enterocytes',
'2': 'Enterocytes',
'3': 'Enteroendocrine',
'4': 'Enteroendocrine progenitor',
'5': 'Tuft cells',
'6': 'TA cells',
'7': 'TA cells',
'8': 'Stem cells',
'9': 'Paneth cells',
'10': 'Goblet cells',
'11': 'Stem cells',
}
adata.obs['cell_type'] = adata.obs.som_cluster_id.map(cell_mapper).astype('str')
adata.write(dataset, compression='gzip')
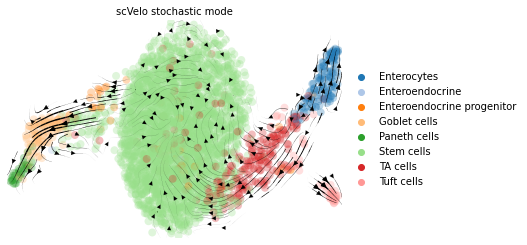
scVelo stochastic
[5]:
title = 'scVelo stochastic mode'
adata.uns['datapath'] = dataset
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=2000)
scv.pp.moments(adata, n_pcs=30, n_neighbors=30)
scv.tl.velocity(adata, mode='stochastic')
scv.tl.velocity_graph(adata)
adata.uns['cell_type_colors'] = ['#1f77b4', '#aec7e8', '#ff7f0e', '#ffbb78', '#2ca02c', '#98df8a', '#d62728', '#ff9896']
scv.pl.velocity_embedding_stream(adata, color=label, title=title, legend_loc='far right')
[5]:
scv.pp.neighbors(adata)
adata_velo = adata[:, adata.var.loc[adata.var['velocity_genes'] == True].index]
exp_metrics["model_dyn"] = utv.evaluate(adata_velo, cluster_edges, label, 'velocity')
# Cross-Boundary Direction Correctness (A->B)
{('Stem cells', 'TA cells'): -0.09643490177153952, ('Stem cells', 'Goblet cells'): 0.19285331884716267}
Total Mean: 0.048209208537811576
# In-cluster Coherence
{'Enterocytes': 0.8242295, 'Enteroendocrine': 0.9842765, 'Enteroendocrine progenitor': 0.87248236, 'Goblet cells': 0.8201039, 'Paneth cells': 0.839217, 'Stem cells': 0.7775167, 'TA cells': 0.8600152, 'Tuft cells': 0.9146982}
Total Mean: 0.8615673780441284
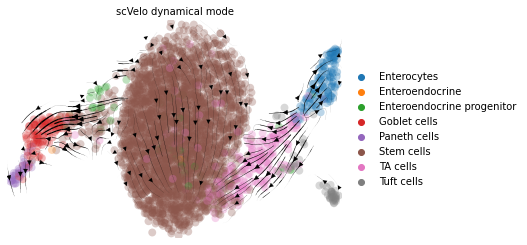
scVelo dynamic
[6]:
title = 'scVelo dynamical mode'
adata.uns['datapath'] = dataset
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=2000)
scv.pp.moments(adata, n_pcs=30, n_neighbors=30)
scv.tl.recover_dynamics(adata, n_jobs=20)
scv.tl.velocity(adata, mode='dynamical')
scv.tl.velocity_graph(adata)
scv.pl.velocity_embedding_stream(adata, color=label, title=title, legend_loc='far right')
[7]:
scv.pp.neighbors(adata)
adata_velo = adata[:, adata.var.loc[adata.var['velocity_genes'] == True].index]
exp_metrics["model_dyn"] = utv.evaluate(adata_velo, cluster_edges, label, 'velocity')
# Cross-Boundary Direction Correctness (A->B)
{('Stem cells', 'TA cells'): -0.05054847599892724, ('Stem cells', 'Goblet cells'): 0.1815118434010139}
Total Mean: 0.06548168370104332
# In-cluster Coherence
{'Enterocytes': 0.7199606522659602, 'Enteroendocrine': 0.9669398810968225, 'Enteroendocrine progenitor': 0.843149997812989, 'Goblet cells': 0.6769492431605317, 'Paneth cells': 0.6947521817913263, 'Stem cells': 0.7846342928623831, 'TA cells': 0.8228564298220513, 'Tuft cells': 0.9238745465967951}
Total Mean: 0.8041396531761074
[7]:
scv.tl.latent_time(adata)
scv.pl.scatter(adata, color='latent_time', color_map='gnuplot', size=20)
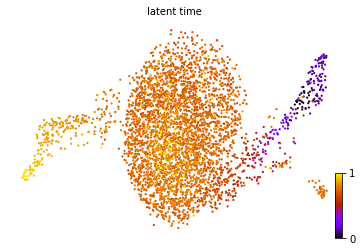
UniTVelo
UniTVelo requires a configuration file as input. You may sub-class it from base config file config.py and override the parameters you need to change (demonstrated below). For the details and comments of each parameter, please refer to config.py.
IROOT represents how cells time should be initialized, default None (based on the exact order of input expression matrix).velo_config.IROOT = None works unsatisfactorily, we recommend change IROOT parameter or alternatively, set velo_config.NUM_REP = 2 to enabel multiple initializations.[8]:
velo_config = utv.config.Configuration()
velo_config.R2_ADJUST = True
velo_config.IROOT = 'Stem cells'
velo_config.FIT_OPTION = '1'
[10]:
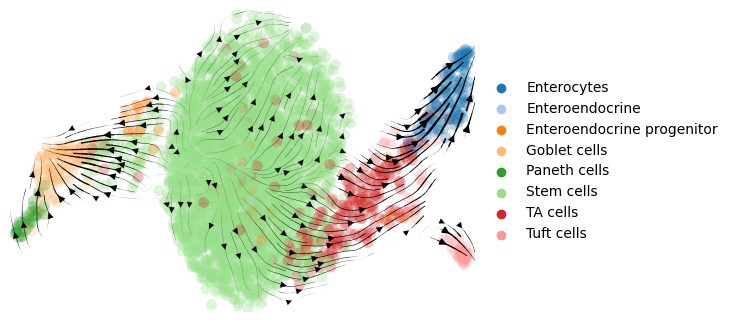
adata = utv.run_model(dataset, label, config_file=velo_config)
adata.uns['cell_type_colors'] = ['#1f77b4', '#aec7e8', '#ff7f0e', '#ffbb78', '#2ca02c', '#98df8a', '#d62728', '#ff9896']
scv.pl.velocity_embedding_stream(adata, color=adata.uns['label'], dpi=100, title='',
legend_loc='far right')
-------> Model Configuration Settings <-------
GPU: 2 FIG_DIR: ./figures/ BASE_FUNCTION: Gaussian
GENERAL: Curve BASIS: None N_TOP_GENES: 2000
OFFSET_GENES: False FILTER_CELLS: False EXAMINE_GENE: False
RESCALE_TIME: False RESCALE_DATA: True R2_ADJUST: True
IROOT: Stem cells NUM_REPEAT: 1 FIT_OPTION: 1
DENSITY: SVD REORDER_CELL: Soft_Reorder AGGREGATE_T: True
ASSIGN_POS_U: False WIN_SIZE: 50 LEARNING_RATE: 0.01
MAX_ITER: 10000 USE_RAW: False RAW_GENES: False
---> # of velocity genes used 433
---> # of velocity genes used 414
---> # of velocity genes used 414
---> Use Diffusion Pseudotime as initial.
[6]:
scv.pp.neighbors(adata)
adata_velo = adata[:, adata.var.loc[adata.var['velocity_genes'] == True].index]
exp_metrics["model_dyn"] = utv.evaluate(adata_velo, cluster_edges, label, 'velocity')
# Cross-Boundary Direction Correctness (A->B)
{('Stem cells', 'TA cells'): 0.31262964284932904, ('Stem cells', 'Goblet cells'): 0.8771414068365261}
Total Mean: 0.5948855248429276
# In-cluster Coherence
{'Enterocytes': 0.9587416014203398, 'Enteroendocrine': 0.996026293460715, 'Enteroendocrine progenitor': 0.9934198592433778, 'Goblet cells': 0.9895047323373496, 'Paneth cells': 0.9921224640938455, 'Stem cells': 0.9917418236918903, 'TA cells': 0.9589615744200212, 'Tuft cells': 0.9773175395058773}
Total Mean: 0.982229486021677
[7]:
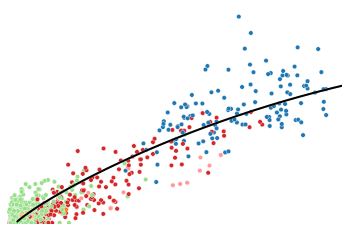
scv.tl.latent_time(adata, min_likelihood=None)
scv.pl.scatter(adata, color='latent_time', color_map='gnuplot', size=20)

\(R^2\) for Informative Genes
[7]:
subvar = adata.var.loc[adata.var['velocity_genes'] == True]
sub = adata[:, subvar.index]
[9]:
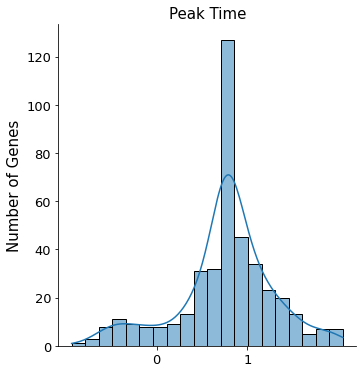
import seaborn as sns
import matplotlib.pyplot as plt
sns.displot(sub.var['fit_t0'].values, kde=True, bins=20)
plt.xticks([0, 1], fontsize=13)
plt.yticks(fontsize=13)
plt.ylabel('Number of Genes', fontsize=15)
plt.title('Peak Time', fontsize=15)
[9]:
Text(0.5, 1.0, 'Peak Time')
[8]:
r2 = sub.var[['fit_t0', 'fit_sr2', 'fit_ur2']].sort_values(by=['fit_sr2'], ascending=False)
r2
[8]:
| fit_t0 | fit_r2 | |
|---|---|---|
| index | ||
| Dgat1 | 0.773396 | 0.903234 |
| Gsta4 | 0.953815 | 0.887514 |
| Cyp2c66 | 0.763647 | 0.886013 |
| Cyp2c65 | 0.761451 | 0.878404 |
| Slc2a2 | 0.752518 | 0.873184 |
| ... | ... | ... |
| Edn1 | 1.871733 | -0.015091 |
| Tmem176b | 0.774602 | -0.017087 |
| Gnai2 | 1.330305 | -0.037739 |
| Chgb | 1.595687 | -0.038026 |
| Krt18 | 1.449424 | -0.128269 |
414 rows × 2 columns
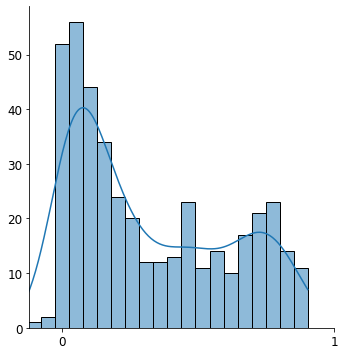
[14]:
sns.displot(r2['fit_sr2'].values, kde=True, bins=20)
plt.xlim([-0.12, 0.89])
plt.xticks([0, 1], fontsize=12)
plt.yticks(fontsize=12)
plt.xlabel('')
plt.ylabel('')
[14]:
Text(10.049999999999997, 0.5, '')
[12]:
utv.pl.plot_range('Gsta4', adata, velo_config, show_legend=False, show_ax=False)
[13]:
utv.pl.plot_range('Atp2a3', adata, velo_config, show_legend=False, show_ax=False)

[11]:
gene_name = 'Muc13'
adata.obs['temp'] = adata[:, gene_name].layers['Ms']
scv.pl.scatter(adata, color='temp', color_map='viridis', size=20)
Detailed clusters
[ ]:
cell_mapper = {
'1': 'Enterocytes',
'2': 'Enterocytes',
'3': 'Enteroendocrine',
'4': 'Enteroendocrine progenitor',
'5': 'Tuft cells',
'6': 'TA cells',
'7': 'TA cells',
'8': 'Stem cells',
'9': 'Paneth cells',
'10': 'Goblet cells',
'11': 'Stem cells',
}
adata.obs['cell_type'] = adata.obs.som_cluster_id.map(cell_mapper).astype('str')
[ ]:
scv.pl.velocity_embedding_stream(adata, color='cell_type', dpi=300, legend_loc='far right', title='', save='Organoids_Generalized Option 1_Detailed.png')
[ ]:
cluster_edges = [
("Stem cells", "Goblet cells"),
("Stem cells", "TA cells"),
("Enteroendocrine progenitor", "Goblet cells")]